單體聚合的熱力學(xué)
單體的熱力學(xué)決定著單體能否進(jìn)行聚合,也就是說決定于在聚合反應(yīng)中能量互相轉(zhuǎn)化各因素,。從熱力學(xué)觀點(diǎn)來看,,聚合反應(yīng)中分子量較小的單體變成分子量很高的聚合物時(shí),體系中的自由能將減少,,即△F為負(fù)值時(shí)才能進(jìn)行聚合,。而自由能的變化又是與熱焓及熵的變化相關(guān)聯(lián)的。

所以,,測定聚合時(shí)熱始H和嫡S的變化就可以得到自由能的數(shù)值,。
聚合反應(yīng)中熱始的変化,可通過反應(yīng)前后鍵能的變化求出(理論值),,也可由參加聚合單體反應(yīng)前后生成熱的變化求出,。因有聚合能力的單體都含有雙鍵,聚合后雙鍵打開,,π鍵變成α鍵,。

這種熱焓的變化對聚合有利。而熵的變化恰好相反,,因單體變?yōu)榫酆衔飼r(shí)分子數(shù)目減少,,熵值變大(S=KI.nW),因此聚合時(shí)體系中嫡變△S為負(fù)值,,引起△F值增大,,不利于聚合。聚合時(shí)的熵變減少數(shù)值約為25~30卡/克分子?K,。在298°K時(shí),,TAS約為7.5~9千卡/克分子。所以只有在聚合時(shí)放出的熱△H大于7,,5~9千卡/克分子時(shí),,オ能使體系中自由能減少,聚合反應(yīng)才能進(jìn)行,。理論上,,當(dāng)某種單體的聚合熱小于7,5~9千卡/克分子時(shí),,就不能聚合成高聚物,。
其它
1.轉(zhuǎn)化率的變化
聚合反應(yīng)是很復(fù)雜的反應(yīng)過程,根據(jù)游離基聚合反應(yīng)動(dòng)力學(xué)可得到游離基聚合反應(yīng)初期的聚合速度方程式,,由該方程式得知引發(fā)劑引發(fā)聚合的速度與引發(fā)劑濃度的平方根成正比,。但在實(shí)際上根據(jù)這一點(diǎn)來控制反應(yīng)速度又有發(fā)生困難,其原因可作期如下解釋。
在聚合反應(yīng)整個(gè)過程中,,聚合速度是不斷變化的,,如一些單體(氯乙烯、苯乙烯,、甲基丙烯酸甲酯等)在間歇式聚合時(shí),,隨著轉(zhuǎn)化率的逐漸增大,聚合速度也發(fā)生變化,。轉(zhuǎn)化率隨聚合反應(yīng)時(shí)間變化的典型的曲線常呈S型,,如圖1-1所示。聚合反應(yīng)可分為四個(gè)階段,。
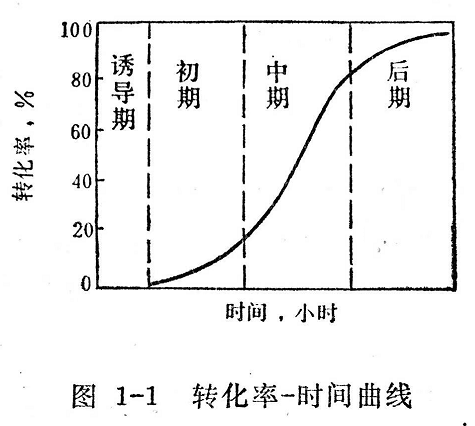
誘導(dǎo)期:此時(shí)轉(zhuǎn)化率為零,,沒有聚合物生成,聚合速度也為零(因體系中存在著阻聚作用),。
聚合初期:通常指轉(zhuǎn)化率在20%以下,,這段時(shí)期聚合速度較平穩(wěn)。
聚合中期:以氯乙烯為例:在轉(zhuǎn)化率達(dá)到50%左右時(shí),,隨轉(zhuǎn)化率的增加,,體系的粘度增大,使活性鏈活動(dòng)能力受到阻礙,,鏈終止機(jī)會(huì)減少,,活性鏈積累數(shù)目增多。同時(shí)又因單體量大,,活動(dòng)也較易,,因此聚合反應(yīng)速度自動(dòng)加快,聚合度也隨之增加,。這種聚合速度自動(dòng)加快的現(xiàn)象稱為“自動(dòng)加速效應(yīng)”,。因?yàn)槭窃隗w系粘度增大后出現(xiàn)的,習(xí)慣上又稱為“凝膠效應(yīng)”,。
聚合后期:轉(zhuǎn)化率在50%以上時(shí),,體系粘度更大,鏈終止機(jī)會(huì)更少,。因轉(zhuǎn)化率很高,單體濃度下降較多,,鏈增長機(jī)會(huì)減少,,聚合速度也就逐漸下降。由于在反應(yīng)后期向聚合物轉(zhuǎn)移的機(jī)會(huì)增多,,易產(chǎn)生支化或交聯(lián),。
2.動(dòng)力學(xué)鏈長與平均聚合度
高聚物分子量大小是一個(gè)重要的控制指標(biāo),聚合物必須達(dá)到一定分子量后才具有足夠的強(qiáng)度,。分子量過低,,不僅強(qiáng)度差,,熱穩(wěn)定性也差。不過分子量太高又會(huì)使加工成型困難,。
游離基聚合反應(yīng)中所得產(chǎn)物的分子量是不均一的,。因?yàn)楦鞣N鏈長不等的活性鏈的終止機(jī)會(huì)有一定的幾率分布,所得產(chǎn)物的分子量大小不等,。所說高聚物的分子量是指平均分子量,,聚合度也是指平均聚合度。
動(dòng)力學(xué)鏈長的定義是每個(gè)鏈游離基從生(鏈引發(fā))到滅(鏈終止)的增長過程中,,每個(gè)活性鏈上具有的單體分子數(shù),。也就是說動(dòng)力學(xué)鏈長就是鏈游離基在鏈終止前的平均鏈長。設(shè)動(dòng)力學(xué)鏈長為,,則:

當(dāng)處于穩(wěn)定態(tài)時(shí),,即(Mつ的増長和消耗的速度相等時(shí)(Vi=Vt),由公式推導(dǎo)得知?jiǎng)恿W(xué)鏈長與聚合速度(鏈增長速度)成反比,,聚合速度愈快時(shí),,動(dòng)力學(xué)鏈長愈短,即聚合物平均分子量愈小,。同時(shí),,動(dòng)力學(xué)鏈長與引發(fā)劑濃度的平方根成反比,引發(fā)劑用量愈多,,則鏈長愈短,,亦即平均分子量愈小。當(dāng)然也還應(yīng)考慮到鏈終止的方式以及鏈轉(zhuǎn)移反應(yīng)的情況,。